昨年、ACS Omega誌に掲載した拘束誘導推論法(restraint-guided inference)の論文[1]では、 Boltz-1上でリガンドの立体化学を改善する手法を提案しました。 あれから半年以上が経ち、 Boltz-2[2]だけでなく、Protenix-v1/v2[3][4]やOpenFold3[6]といった新しいモデルが次々と登場しています。 そこで今回は、これらの最新モデルに対して改めてベンチマークを行い、 リガンドの立体化学に関する問題がどの程度改善されているのかを調べてみました。
比較条件
以下の6条件で比較を行いました。
- AlphaFold3
- Boltz-2(vanilla)
- Boltz-2 + conformer restraints(我々の手法を適用)
- Boltz-2 w/ potential(Boltz-2組み込みのinference-time potentials[2]を使用)
- Protenix-v1
- Protenix-v2
評価データセットは前回の論文と同じPLINDERベースのキラル化合物セット[5]を使用しています。 なお、training cutoff dateについてですが、 Protenix-v1/v2はAF3と同じ2021年9月30日をcutoffとしているため、 Before/After splitによる汎化性能の評価が可能です。 一方、Boltz-2のcutoffは2023年6月1日と新しく、今回のBefore/After splitとは対応しません。 したがって、Boltz-2のAfterの成績がよく見えたとしても、 それが学習データに含まれている構造による恩恵である可能性があり、純粋な比較とは言えない点にご注意ください。
Boltz-2 + conformer restraints については、新たにBoltz-2に対して拘束誘導推論法を実装したレポジトリ (https://github.com/cddlab/boltz_restr) のコードに基づいてベンチマークをとっています。 こちらのコードでは、距離拘束による構造変化の誘導や、 ligand-protein間の拘束(VdW拘束)を新たに実装しています。
なお、OpenFold3[6]については今回のベンチマークに含めていません。 2026年3月にはpreview2のアップデートとともに学習データの全面公開も行われており、 再現性と拡張性の観点から注目度の高いプロジェクトですが、 諸事情により今回のベンチマークに含める事ができませんでした。 今後のベンチマークに含めたいと考えています。
成功率
まず、構造予測が正常に完了し、リガンドのジオメトリが大きく壊れていない(結合が切れる、トポロジーが変わってしまう等がない)かを評価しました。全エントリに対する成功した数の割合を、成功率 (%) で表しています。
| モデル | 成功率 (%) |
|---|---|
| AlphaFold3 | 99.86 |
| Boltz-2 w/ potential | 99.67 |
| Boltz-2 | 99.50 |
| Boltz-2 + conf restr | 99.62 |
| Protenix-v1 | 99.12 |
| Protenix-v2 | 94.73 |
ほとんどのモデルで99%以上の成功率が得られましたが、 Protenix-v2ではリガンドの結合が伸びすぎてトポロジーが崩れてしまうケースが約5%発生しており、やや気になるところです。
キラリティの再現率
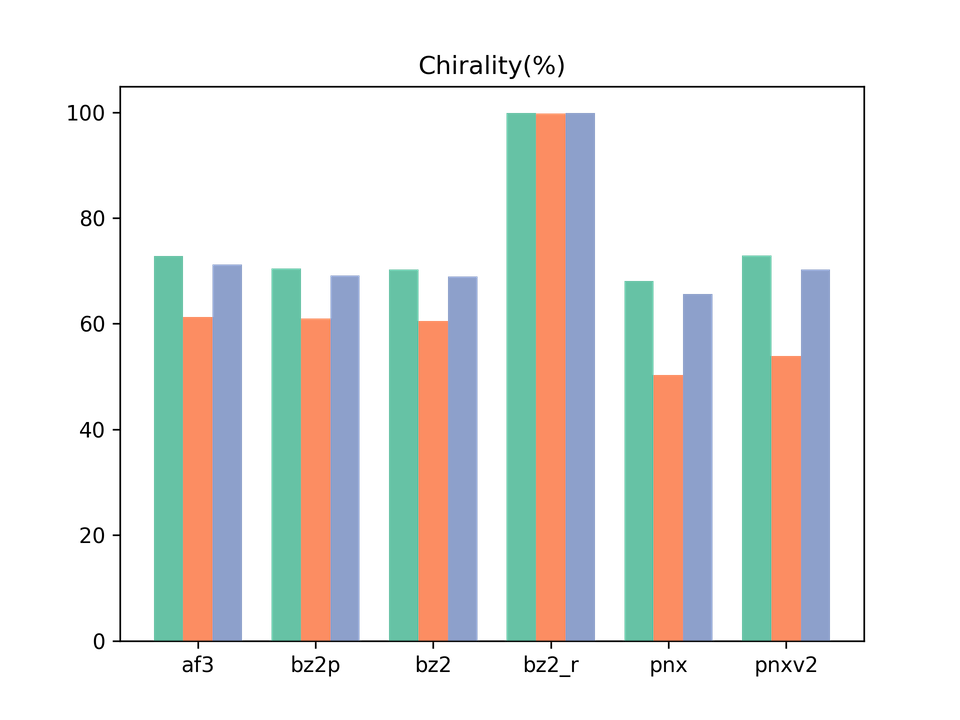
グラフの凡例:af3 = AlphaFold3、bz2p = Boltz-2 w/ potential、bz2 = Boltz-2 (vanilla)、bz2_r = Boltz-2 + conformer restraints、pnx = Protenix-v1、pnxv2 = Protenix-v2。各グループの3本の棒はそれぞれBefore / After / Allに対応します。
前回の論文で注目した指標の一つです。 結果として、Boltz-2 + conformer restraintsのみが100%のキラリティ再現率を達成しました。 これは前回のBoltz-1での結果と一貫しており、我々の手法が新しいモデルベースでも有効であることが確認できました。
一方、Boltz-2のinference-time potentialを使用した条件では、キラリティの改善はあまり見られませんでした。 potentialが本当に正しく効いているのか、何か追加の設定が必要なのかは要検証です。
やや意外だったのは、Protenix-v2でv1よりもキラリティの再現率がわずかに悪化していた点です。 モデルが大きく改善されているにもかかわらず、この点は改善されていないようです。
タンパク質構造の精度 (Protein RMSD)
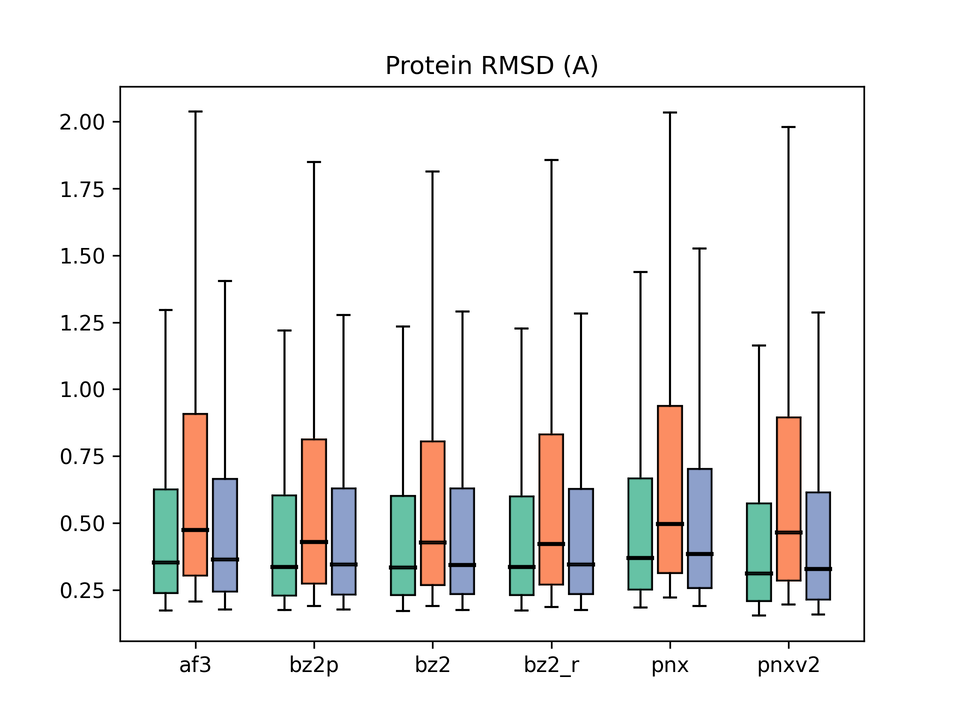
グラフの凡例:af3 = AlphaFold3、bz2p = Boltz-2 w/ potential、bz2 = Boltz-2 (vanilla)、bz2_r = Boltz-2 + conformer restraints、pnx = Protenix-v1、pnxv2 = Protenix-v2。各グループの3本のbox plotはそれぞれBefore / After / Allに対応。
今回の主目的ではありませんが、タンパク質構造の精度も参考として評価しました。 Boltz-2が最も低いmedian RMSDを示し、Before/Afterの乖離も小さい結果でした。 Protenix-v2はv1に比べて改善が見られるものの、Before/Afterの差がやや大きく、 軽度のoverfittingの傾向があるかもしれません。 ただし、全体としてどのモデルも十分に良い精度であり、 差はあるといっても小さく統計的な有意性があるかは不明です。
リガンド結合ポーズの精度(Ligand RMSD)
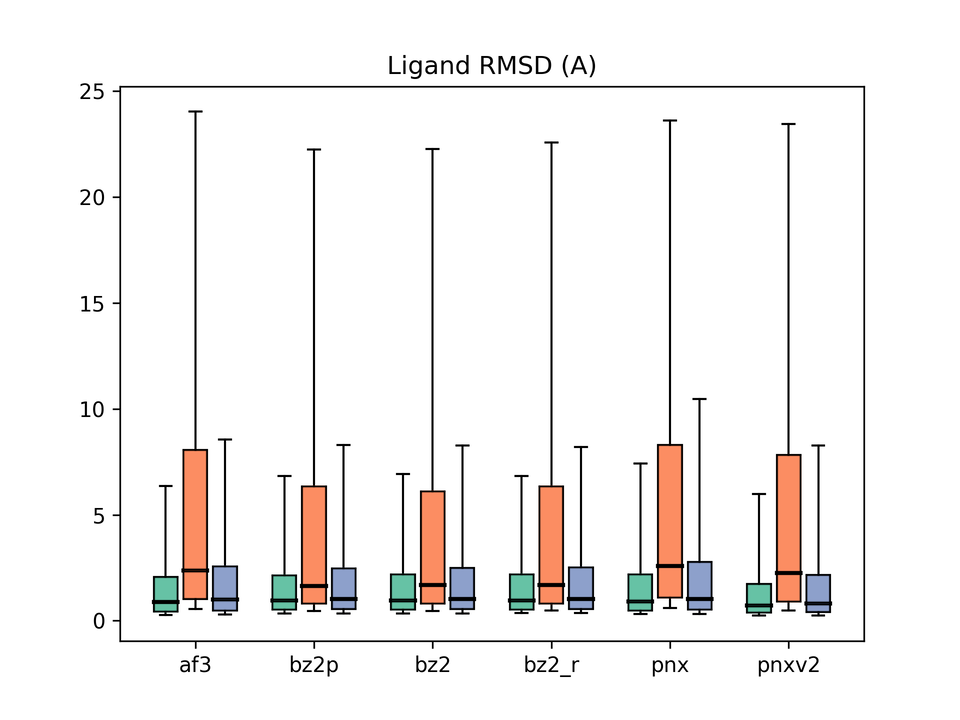
グラフの凡例:af3 = AlphaFold3、bz2p = Boltz-2 w/ potential、bz2 = Boltz-2 (vanilla)、bz2_r = Boltz-2 + conformer restraints、pnx = Protenix-v1、pnxv2 = Protenix-v2。各グループの3本のbox plotはそれぞれBefore / After / Allに対応。
次は、リガンド結合ポーズの予測精度になります。 All(全体)とBefore(training cutoff以前)ではどのモデルも概ね良好ですが、 After(cutoff以降)で差が出ています。
Boltz-2が最も良いmedian Ligand RMSDを示しました。 ただし前述のとおり、これはtraining cutoffの違いによる可能性があります。 AF3は意外にもあまり良くない結果でした。 Protenix-v1が最も悪いスコアでしたが、v2で改善しています。 ただし、v2はAfter/Beforeの差が大きくなっており、過学習の傾向が示唆されます。
リガンドジオメトリの精度(Bond RMSD, Angle RMSD)
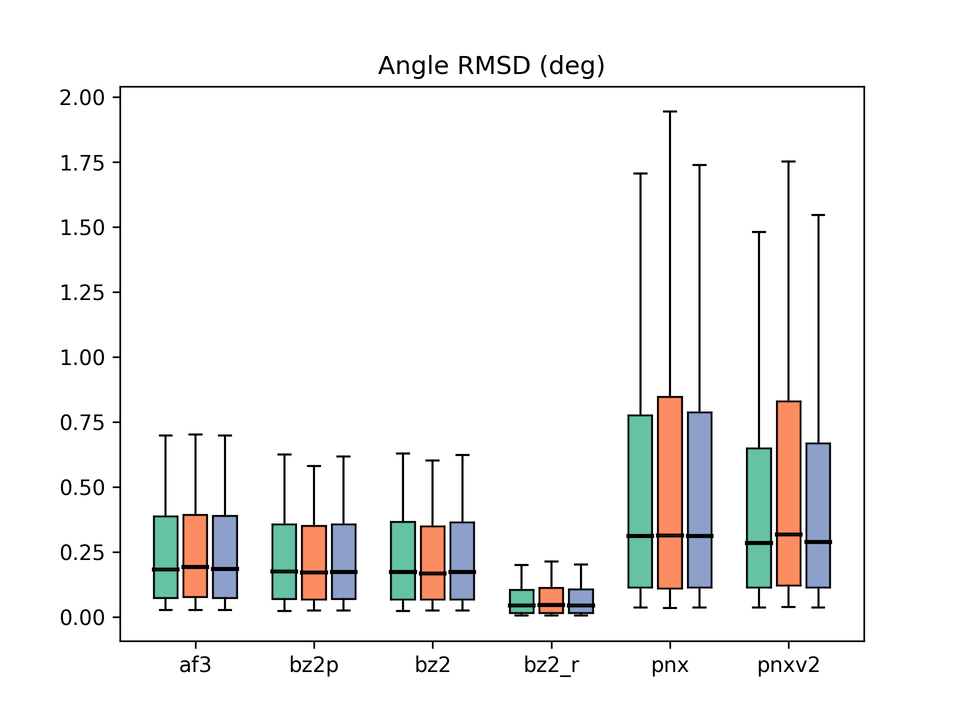
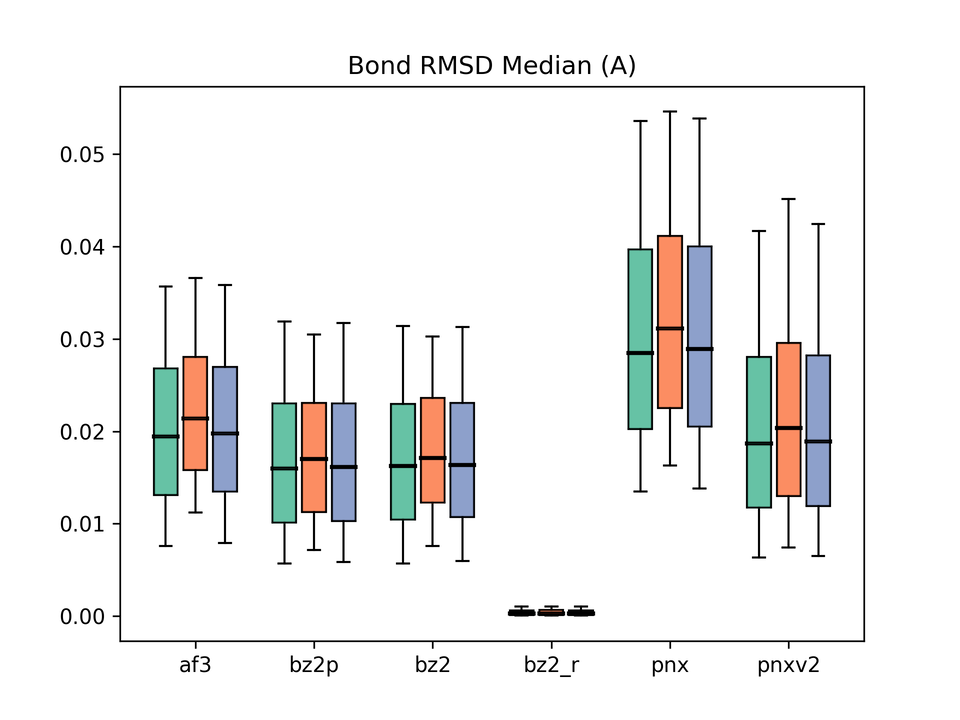
グラフの凡例:af3 = AlphaFold3、bz2p = Boltz-2 w/ potential、bz2 = Boltz-2 (vanilla)、bz2_r = Boltz-2 + conformer restraints、pnx = Protenix-v1、pnxv2 = Protenix-v2。各グループの3本のbox plotはそれぞれBefore / After / Allに対応。
これも、前回の論文で注目した指標の一つです。 リガンドの結合長・結合角の精度についても、Boltz-2が最も良い結果を示しました。 Boltz-2のpotentialを有効にしても、これらの指標にはほとんど改善が見られませんでした。 やはりpotentialが正しく作用しているか疑問が残ります。
一方、conformer restraintsを適用すると劇的に改善されるのは、Boltz-1のときと同様です。 拘束誘導推論法の有効性が改めて確認されました。
Protenix系はリガンドジオメトリ全般に課題がありそうです。 興味深いことに、v2ではBond RMSDについては改善が見られましたが、 Angle RMSDはほとんど改善していないように見えます。 なぜこのような結果になっているかは、やや気になるところです。
タンパク質-リガンド間の最小原子距離
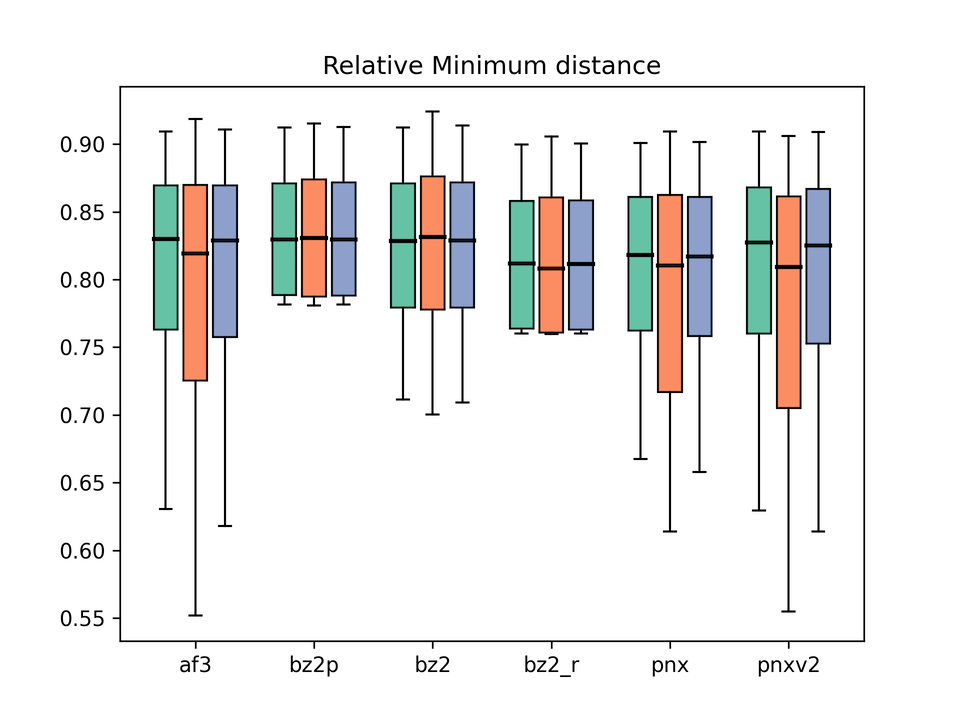
グラフの凡例:af3 = AlphaFold3、bz2p = Boltz-2 w/ potential、bz2 = Boltz-2 (vanilla)、bz2_r = Boltz-2 + conformer restraints、pnx = Protenix-v1、pnxv2 = Protenix-v2。各グループの3本のbox plotはそれぞれBefore / After / Allに対応。
タンパク質とリガンドが物理的にめり込んでいないか(steric clashがないか)を評価する指標です。Posebustersで使われる原子間の相対距離(距離をVdW半径で割って正規化したもの)を評価し、予測構造中での最小値をプロットしています。1だとVdW相互作用している距離と大体同じになるわけですが、水素結合を形成していたりすると1より小さくなる可能性があります。大体0.7以下になっていると近すぎる(めり込んでいる)と見て良いと思います。
結果を見ると、Boltz-2は素の状態(bz2)でもかなり優秀ですが、 potentialを有効にする(bz2p)とめり込みがほぼゼロになることがわかりました。 この点ではpotentialの効果がはっきりと出ていると言えるでしょう。 Boltz-2 with conformer restraints (bz2_r)も同様に、めり込みはほぼゼロでした。 VdW拘束の効果が出ていることがわかります。
対照的に、AF3はsteric clashが多く見られます。 Protenix-v1/v2も同様にめり込みが多く、v2でも改善は見られませんでした。
まとめと今後の展望
今回のベンチマークの主な知見をまとめます。
総合的に見ると、Boltz-2が最も良い結果を示しました。 この結果だけを信じると、cofoldingなどの用途にはBoltz-2(+ inference-time potential / conformer restraints)を用いておくのがベストのようです。 ただし、training cutoff dateの違いがあるため、純粋な性能比較とは言えない点に注意が必要です。 Boltz-2が新しい構造データまで学習に含めているために、見かけ上の性能が向上している可能性があります。
キラリティの再現については、依然としてconformer restraintsの適用が最も効果的です。
Boltz-2のinference-time potential (--use_potentials option)では、steric clashの解消には効果があるものの、
キラリティやリガンドジオメトリの改善にはあまり寄与していないようでした。
この点については検証が必要でしょう。
一方で、Protenix-v2も着実に改善が進んでいるように見えます。 本家のテクニカルレポート[4]でも言及されているように、 少なくともv1よりは良くなっています。 ただ、AF3やBoltz-2と比較してどの程度優れているかは、さらなる検討が必要です。
なお、Protenix-v2にはTraining-Free Guidance(TFG)モジュール[4]が実装されており、 推論時にジオメトリや物理的制約を適用する機能が利用可能なようです。 今回は使い方の詳細が十分に把握できなかったため試すことができませんでしたが、 我々のconformer restraintsと類似のアプローチである可能性があり、 今後検証してみたいと考えています。
本記事は2026年4月時点の研究成果に基づいています。
参考文献
[1] Ishitani, R.; Moriwaki, Y. Improving Stereochemical Limitations in Protein-Ligand Complex Structure Prediction. ACS Omega 2025, 10, 45, 43857–43869.
[2] Passaro, S.; et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv 2025, 2025.06.14.659707.
[3] ByteDance AML AI4Science Team; et al. Protenix - Advancing Structure Prediction Through a Comprehensive AlphaFold3 Reproduction. bioRxiv 2025, 2025.01.08.631967.
[4] ByteDance AML AI4Science Team; et al. Protenix-v2: Broadening the Reach of Structure Prediction and Biomolecular Design. bioRxiv 2026, 2026.04.10.717613.
[5] Durairaj, J.; et al. PLINDER: The Protein-Ligand Interactions Dataset and Evaluation Resource. bioRxiv 2024, 2024.07.17.603955.
[6] The OpenFold3 Team. OpenFold3-preview. GitHub, 2025. https://github.com/aqlaboratory/openfold-3