准教授の森脇です。最近弊ラボから出版された生合成遺伝子クラスター(BGC)内の網羅的なタンパク質複合体予測の論文を紹介します。
ざっくり言えば、より高速に複合体予測を回せるようにしたAlphaFold3を用いて、BGCの中に存在する興味深いヘテロ複合体を網羅的に探索したという論文です。
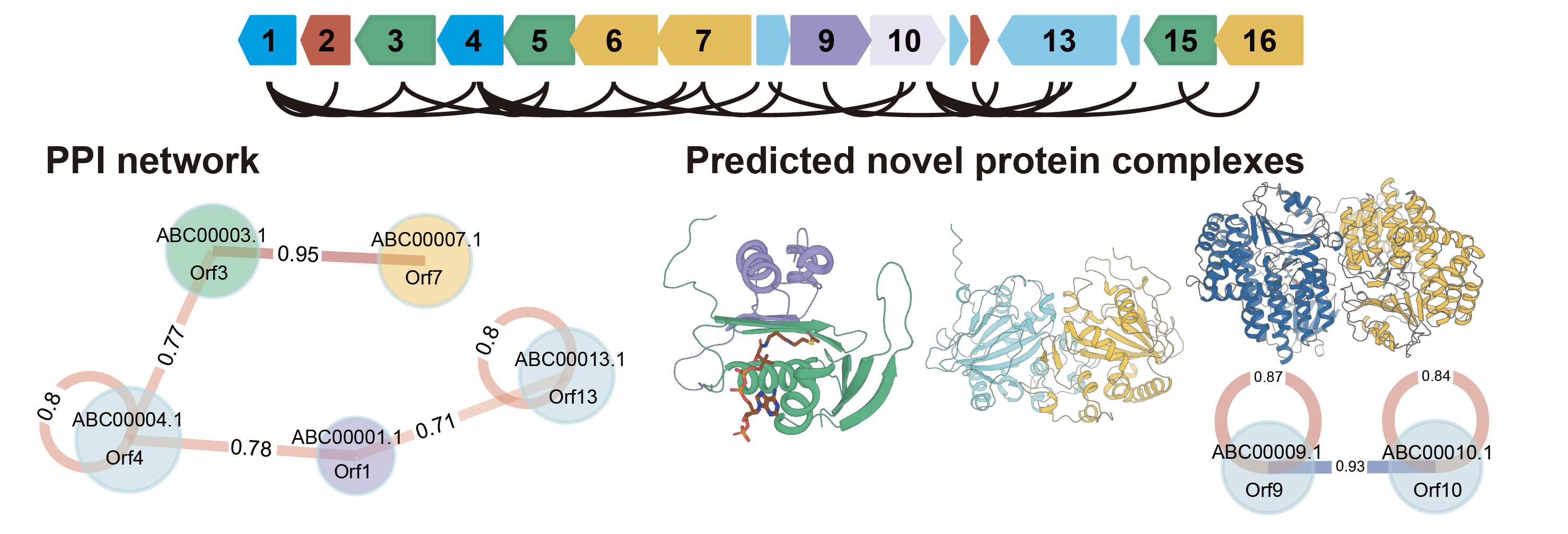
BGCの中には、PKSやNRPSのコアモジュールとして研究されているタイプのタンパク質もあれば、BGC外のタンパク質とのアミノ酸配列比較情報を用いてもその機能がまったく予測できず”hypothetical protein”とだけラベリングされているタンパク質も存在しています。一方、いくつかの実験的な先行研究によれば、BGCの”hypothetical protein”の中には2つ(以上)のタンパク質が揃うことで初めてある1つの機能を発現させる例が示されていました。このように、タンパク質の中には複合体を形成することでその機能を発揮するタイプのタンパク質も存在し得ますが、それらの機能は配列解析から予測することは本質的に困難です。本研究では2つ以上がセットになって初めて機能するタンパク質の複合体を網羅的に予測することで、従来法では困難とされていた、BGCにコードされているタンパク質間の新しい相互作用ネットワークの理解につながることを示しました。
本研究の成果は Computational and Structural Biotechnology Journal誌に掲載されました。
研究背景:生合成遺伝子クラスター(BGC)の話と機能予測
二次代謝産物は、生物の成長・発生・繁殖に直接関与しない天然物です。アミノ酸・核酸・糖といった一次代謝産物とは異なり、捕食者に対する防御1、他生物との競合2、共生関係3といった生態学的相互作用において特化した役割を担うことが多いのですが、これらの化合物は通常、特定の代謝産物の生合成に必要な酵素群や、トランスポーター・調節タンパク質などの補助タンパク質をコードする遺伝子が一カ所に集まった生合成遺伝子クラスター(BGC)によって産生される4,5。非リボソームペプチド(NRP)、ポリケチド(PK)、リボソーム合成後修飾ペプチド(RiPP)、テルペン、糖などの多様な化学構造を含むため、酵素学・有機化学の両面から大きな関心を集めてきました。時代が下るにつれ(2000〜2010年頃)、次世代シーケンシングやメタゲノム解析などのシーケンシング技術の進歩、バイオインフォマティクスツールの発展、ゲノムデータベースの拡充により、難培養微生物由来の二次代謝産物研究は大きく前進しました。2020年代頃からはAlphaFold2(AF2)25に代表される高精度な立体構造予測が登場し、antiSMASH6-13やDeepBGC15、ClusterFinder16のような配列ベースの手法だけでは得難いタンパク質間相互作用に関する有用な予測情報を提供できるようになりました。
一般に、タンパク質は他のタンパク質やリガンドと複合体を形成することで機能を拡張できます。というか、他のタンパク質やリガンドと結合しないタンパク質は生体中であまり存在しておらず、通常何らかの形で相互作用を介して機能を発揮しています。AlphaFoldシリーズのタンパク質–タンパク質複合体予測は現状もっとも精度が高く、これを用いた新規な複合体予測からの実験実証が、Nature, Cell, Scienceなどのトップジャーナルでもよく観測されます。BGCにおいても複合体形成は重要であり、キャリアタンパク質17、trans-AT型PKS18、cis-AT型PKS19、I型/II型PKSや脂肪酸合成酵素20,21で観察されるタンパク質間相互作用が詳細に研究されてきました。
本研究では、AlphaFold3(AF3)31を用い、MIBiGデータベースに登録されている2,437個のBGCにコードされるタンパク質間の総当たり複合体予測を網羅的に実施しました。その結果、487,828ペアの中から興味深いホモ二量体・ヘテロ二量体を発見することができました。それらの中には、既知のヘテロ複合体の構造とよく一致するものもあれば、今後の実験的検証が大いに期待される新規複合体も含まれていました。これらの結果は、BGCにコードされる隠れたタンパク質間の相互作用ネットワークを理解し、二次代謝産物の新奇な生合成機構の解明や新規化合物の発見に貢献することが期待されます。
研究手法
対象タンパク質の選定:MIBiG version 4.0(2024-11-15)に登録され、現在も維持・更新されている2,437の「active」なBGCに着目しました。なお、Type I PKS/NRPSに多く見られる1,950アミノ酸を超えるタンパク質は、二量体計算時にGPUで計算しきれないため予測対象から除外しています。
MSA生成と構造予測:高精度かつ高速な予測を両立するため、AF3標準のHMMER 332によるMSA生成パイプラインを、LocalColabFold33のcolabfold_searchコマンドで置き換えました。このMSA変換部分の処理は我々が開発したalphafold3_toolsのaf3tools msatojsonコマンドで利用可能です。
予測複合体の検証:生物学的に妥当な(biologically-relevantな)相互作用を同定するため、ipTMとipSAE35の2つの指標を用いました。ipTMはサブユニット間の相対配置の精度を測り、≥ 0.8で高信頼、< 0.6で予測失敗、0.6〜0.8は不確実領域とされます。また、ipSAE_min36、pDock37、pDockQ238、LIS39も提供しています。MSA生成法の影響を評価するため、MMseqs2/alphafold3_toolsワークフローとHMMER3ベースの生成を、4つのBGC由来の全ペア($n = 1184$)で比較しました。また、PDBに登録された生物学的集合体(biological assembly)と化学量論情報をもとに、配列同一性95%以上の閾値で予測複合体を実験構造と比較検証しました。ヘテロ二量体については、Gemmi version 0.7.340を用いて2サブユニット間の主鎖重原子のRMSDを算出しました(20残基未満は除外)。
この複合体の妥当性の評価については、最近AlphaFold Databaseの”How confident should I be in a predicted complex?”の項に詳細に書かれていますので、興味がある方はご確認下さい。当初はipTMの値だけだったのですが、6月のいつ頃からか、ipSAEやpDockQ2, LISなどの指標も合わせて説明が掲載されています。さらに、これらの評価指標についてはalphafold3_tools ver.0.4.1からaf3tools metricsでまとめて算出できるようになっています。詳細はこちらから。ぜひ使ってみて下さい。
組換えタンパク質の発現・精製:後述のKetosynthase(KS)–Chain Length Factor(CLF)の代表例としてのmbgクラスターを検証するため、pETDuet-1ベクターを用いてE. coli BL21(DE3)で2遺伝子を共発現させ、Ni-NTAアフィニティー精製とSDS-PAGEで複合体形成を評価しました。本論文で唯一実験データのある部分です。
ネットワーク可視化:2,437 BGCのタンパク質間ネットワークをNetworkX version 3.5で描画しました。結果は(http://vivace.bi.a.u-tokyo.ac.jp/network_in_bgc/publish.html)に掲載してあります。
結果
高速複合体予測パイプライン
MIBiGの「active」な2,437 BGCを対象に、最大1,950残基のタンパク質について全組み合わせ(自己ペアを含む)の複合体を予測しました。$N$個のタンパク質を含むBGCでは$N(N+1)/2$ペアの予測を行うことになります($N$個のタンパク質に対し、$N$個のホモダイマー予測と$N(N-1)/2$のヘテロダイマー予測を足したもの)。MMSeqs2と768 GB以上のRAMを持つサーバー(今回は主に名古屋大学の不老)で1複合体あたり1〜2分でMSAを生成し、計487,828ペアの構造予測を行いました。実際には合計2000残基以内をRTX4090や5090に、3950残基をTSUBAME 4.0のH100 GPU計算機に分散して計算させました。
MMseqs2/alphafold3_toolsとHMMER3の予測を1,184複合体で比較したところ、両者は良く一致しました。特にipSAE、ipSAE_min、pDockQ2、LISは相関係数0.90〜0.93を示しました(Figure 2)。これらの指標はipTMやpDockQと異なり多くの複合体で0.0となり、その多くはipTM・pDockQも低い複合体と重なりました。これらの観察から、本研究ではipSAEとipTMを主要な信頼度指標として用いました。一方、MSA生成法によって信頼度指標が大きく異なる例も観察され(例:BGC0000001のAEK75492.1–AEK75516.1ではMMseqs2でipTM=0.79、HMMER3でipTM=0.11)、1,184予測中ipTMで71ペア(6.0%)、ipSAEで23ペア(1.9%)が0.2を超える差を示しており、MSA生成法の選択が予測信頼度に影響し、偽陰性・偽陽性につながりうることが示されています。
予測複合体の検証
2024年11月30日時点で、MIBiG ver. 4.0と95%以上の配列同一性を示すPDBエントリは、ホモ多量体555件、ヘテロ多量体30件であった。PDBにホモ二量体として登録されたタンパク質では、予測二量体モデルの72.1%(294/408)がipTM ≥ 0.8、87.7%(358/408)がipTM ≥ 0.6を示しました。ipSAEは0.6以上でipTMと高く相関し、それ以下では0に近づきました。高次のホモ多量体を形成する135タンパク質は二量体として予測するとipTMが0.6付近となることが多かったが、PDBの化学量論で再予測すると多くがipTMが0.8を超え、大幅に改善していました。代表例のCutA1(イネ、PDB ID: 2ZOM)では、二量体予測は信頼度が低かった(ipTM=0.38)が、三量体予測では実験構造を良く再現した(ipTM=0.64、ipSAE=0.84)。さらにN末端の長いトランジットペプチド領域(1–64残基)を除くとipTM=0.93、ipSAE=0.86まで改善し、長い天然変性領域がipTMを低下させること、そしてipSAEがそのような場合でも信頼性の高い指標であることが示されました。
ヘテロ複合体の検証
30のヘテロ複合体でもホモ複合体と同様のipTM–ipSAE相関が見られました。このうちPDB ID: 6M01、7YN3、8K4R、8T19、4TX3はipTM < 0.6を示しました。7YN3・8K4R・6M01では結晶構造取得のために2タンパク質間の共有結合架橋試薬が用いられており、一方がアシルキャリアタンパク質(ACP)です。ACPは特定パートナーと安定な複合体を作るのではなく、複数のパートナー酵素と一過的に相互作用して中間体を運搬・修飾するため、相互作用が弱くipTMが高くならないと考えられます。さらにACP結合基質の化学構造が不明・未登録であることや、AF3のタンパク質–リガンド予測の限界も、一過的なACP複合体の予測を難しくしている。8T19はRiPP前駆体ペプチドPbtAとその修飾酵素PbtFの複合体で、AF3は結晶構造から2残基ずれた位置にペプチドが結合すると予測しました(ipTM=0.39)。RiPP前駆体ペプチド結合タンパク質への理解を深めるため、NisB(前駆体NisAとの結晶構造は同一鎖への融合50または化学修飾51のため検証データから除外)やStrB(SuiB52と94.98%同一)49を調べたところ、予測構造は対応する結晶構造を良く再現しました。総じて、AF3は30のヘテロ複合体のうち26例で複合体形成(ipTM ≥ 0.6)を正しく予測したが、ACPや前駆体ペプチドのような一過的・小界面の複合体には予測能力に限界があることがわかります。
新規ヘテロ複合体
487,828ペア中、15,438のヘテロ二量体がipTM ≥ 0.6、3,754がipTM ≥ 0.8を示し、機能未知タンパク質が形成する複合体も含まれました。borrelidin BGC(BGC0000031)53-55の「hypothetical protein」cae45685.1とcae45686.1のヘテロ二量体は、GCN5関連N-アセチルトランスフェラーゼ(GNAT)と部分的な配列類似性を示し、Foldseek57による構造検索ではヘテロ三量体NatC複合体のNaa30サブユニット58に類似しました。複合体界面にアセチルCoAの結合ポケットが形成され、N末端アセチル化を触媒する可能性が示唆されました。実際、類似BGCを持つと考えられるStreptomyces mutabilis sp. MIIがN-Acetylborrelidin Bを産生すると報告されており59、この複合体がborrelidin Bのアセチル化を触媒する妥当性があります。また、tropodithietic acid BGC(BGC0000932)60のDUF4399ドメイン含有タンパク質PGA1_262p00820は、DoxXファミリーのPGA1_262p00830と複合体を形成すると予測されました(ipTM=0.82)。単量体では天然変性と予測されたN末端領域が、ヘテロ二量体ではα-ヘリックスを形成して界面を作っており、両者が膜結合型の酵素やトランスポーターとして協調的に機能する可能性が示されました。
このように、新規ヘテロ複合体の予測は、これまでDUFとされてきたタンパク質の機能予測や、BGCにコードされるタンパク質間の新しい相互作用ネットワークの理解につながる可能性があります。
構造的に相同なヘテロ複合体
予測ヘテロ二量体のうち、構成タンパク質間の構造RMSDが≤1.0 Åのものが952ペア(4.9%)、≤2.0 Åが1,390ペア(7.1%)あり、後者のipTM・ipSAE中央値は0.82・0.71と高信頼でした。代表例はType II PKSの中心触媒装置であるketosynthase (KS)–chain length factor(CLF)複合体で、actinorhodin BGC(BGC0000194)のKS(CAC44200.1)とCLF(CAC44201.1)はipTM=0.96、ipSAE=0.93で結晶構造(PDB ID: 1TQY)を良く再現しました。両者はRMSD 1.02 Åで同じフォールドを共有しますが、KSのホモ二量体予測は低スコア(ipTM=0.65)、CLFはほぼシグナルなし(ipTM=0.20)であり、AF3が単量体間の構造類似性とは独立にヘテロ複合体形成を正しく識別できることが示されました。
複数のKS・CLFを含むmbgクラスター(BGC0002441)では、既報で実験的に検証された正しいペアリング(Mbg7–Mbg3など)41に対しipTM・ipSAEが最大値を示しました。特にipSAEは誤ったペアでipTMより低い値を示し、識別力が高いことが示されました。共発現実験でもMbg7–Mbg3、Mbg6–Mbg5は共溶出した一方、Mbg7–Mbg5(ipTM=0.83、ipSAE=0.75)は複合体を形成せず、高い指標値でも偽陽性が起こりうることが示されました。同様の傾向はYsfクラスター(BGC0002547)62でも見られました。
ホモ・ヘテロ両方の二量体を形成しうるペアも381組同定されました。代表例のmonensin BGC(BGC0000100)のMonBIとMonBIIでは、MonBI単独では酵素活性を欠くがMonBIIとのヘテロ二量体で活性を示しました63。AF3はMonBI–MonBIIヘテロ二量体を最も高いipTM/ipSAE(0.93/0.88)で予測し、最近決定された結晶構造(PDB ID: 9KW6)64とも良く一致しました。これは両者の二量体形成が競合的で、ヘテロ二量体が優先的に形成されることを示唆しています。armeniaspirol BGC(BGC0002022)65では、フラビン依存性ハロゲナーゼ様タンパク質Ams8・Ams9・Ams22に着目しました。Ams22はPltA(PDB ID: 5DBJ66)と類似しジクロロ化を触媒すると考えられます。Ams8とAms9はそれぞれホモ二量体(ipTM=0.77、0.73)よりヘテロ二量体(ipTM=0.86)で高い値を示しました。Ams8ではFAD結合を阻害する位置にE337があり、また反応に必須の保存されたLys残基67-69がN90に置換されているため、ハロゲナーゼ活性を欠くと考えられます。一方ams8の遺伝子破壊は生合成を完全に消失させてしまうことから、Ams9がトリクロロ化を触媒する酵素、Ams8が複合体形成を介してAms9の活性を支える補助タンパク質として機能することが考えられます。
ネットワークマップの可視化
2,437 BGCのタンパク質間ネットワーク図を可視化するウェブサイト(http://vivace.bi.a.u-tokyo.ac.jp/network_in_bgc/publish.html)を作成しました。MIBiG ID・分類群・クラス・化合物名でフィルタリングでき、産物の代表的な化学構造も閲覧できるにしてあります。弱いながらも意味のある相互作用の見落としを減らすため、ipTM > 0.55という比較的緩い閾値を用いました。ネットワーク図生成用のJSONファイルとPythonスクリプトはZenodo(https://doi.org/10.5281/zenodo.20536372 )で公開し、ユーザーが任意の閾値や指標で再解析できるようにしています。
Discussion・限界点
本研究では、AF3に基づく高速複合体予測パイプラインを構築し、BGC内のタンパク質間相互作用を網羅的に検出できることを示しました。BGCにコードされる多くのタンパク質が、その生物機能のためにホモ・ヘテロ多量体相互作用に依存していることが示唆され、生合成経路の理解における複合体形成の重要性が浮かび上がってきます。
しかし、本手法には注意すべき限界点があります。
- 弱い、またはtransientな複合体を正確に予測できないこと
- 巨大タンパク質に対する複合体予測が不十分であること
- コンフォメーション変化を起こすタンパク質の場合には予測が不正確になること
- ヘテロ3量体以上を予測していないこと
特にACPや前駆体ペプチドの相互作用を正確には予測することは困難でした。これらは生合成の中でも極めて重要で、生成物の骨格を作るうえで重要な役割を果たしている事が多いのですが、これらはおそらくホスホパンテテイン結合部位を介した共有結合性基質とのprotein-ligand相互作用によって安定化されているという側面が大きいため、AF3のタンパク質–リガンド予測の限界もあって、これらの複合体の予測は現状難しいと考えられます。
一方で、ipTMやipSAEのような指標が、複数の類似した複合体候補の中で生物学的に正しいペアリングを見つけることに有用であるということが示されたというのは興味深い結果です。また、ipSAEは現在デザインした人工タンパク質が標的タンパク質と強く結合できるかどうかの計算的な指標(de novo binder prediction)として期待されています。この結果はAF3の機械学習モデルが原子レベルで相互作用を認識できつつあることを想起させるものです。例えばipSAEの値の大小を用いることで、ある生物種の中にある複数の構造類似タンパク質の中から、真に標的タンパク質と結合できるものを選出できるというような応用可能性があると言えます。
データとコードの公開
BGCネットワークマップはウェブサイト(http://vivace.bi.a.u-tokyo.ac.jp/network_in_bgc/publish.html)で公開しています。
本論文で用いていたMSAの変換コマンドやPAEの可視化コマンドはGitHubで公開しています。現在は論文中で紹介した機能だけでなく、様々な便利ツールも取り揃えつつ、PyPIに登録してpipコマンドでインストールできるようにしました。
こちら(https://pypi.org/project/alphafold3-tools/)からインストールできます。ぜひ使ってみて下さい。
あとがき(ポエム)
動機
この研究のそもそものきっかけとなったのは、2022年度〜2026年度で行われている学術変革A「予知生合成科学」に私が計画班研究分担者として参画したことです。2022年の私はAlphaFold2というビッグウェーブが世に出たこともあって世間をPUIPUI言わせて(?)いましたが、当時の所属専攻である東京大学大学院農学生命科学研究科応用生命工学専攻に生合成を専門としている先生が多かったにも関わらずその先生方とほとんどつながりがなかったため、めくるめく天然物生合成の世界に少ししか触れたことがありませんでした。しかし、この学術変革に参画したことで、天然物だけでなく非常に興味深く新規性の高い酵素の発掘と研究を行う先生方に巡り会えたことは私にとって貴重な経験となりました。
研究の着想は、公開シンポジウムと呼ばれる予知生合成科学の集まりの中で、あるBGCの中で非常に新規性のあるヘテロダイマーが発見され、それがその最終生成物に至る上で重要な修飾酵素として働いているというお話を複数の先生から聞いたことに遡ります。それらのタンパク質ペアはAlphaFoldが出る前から発見されていたものもありましたが、AlphaFoldとはすごいもので、そのタンパク質ペアの配列を入れて数分でそのペアが強い複合体を形成するかどうかを判定できます。それならば、「新規性の高いものがAlphaFoldで見つかるなら、最初から計算で全部やってみたらいいのでは?」と思いつき、高速な計算パイプラインを構築しました。論文中には述べていませんでしたが、公開シンポジウム中では各BGCを専門に研究されている先生や学生とお話して、予測されたヘテロ複合体が確かに機能していることを確かめられたということも聞いています。その新規なヘテロ複合体が具体的にどのような酵素反応を触媒しているかまで予測することはまだ残念ながら難しいのですが、実験的な検証を促進するということには大いに貢献できるのではないかと思います。
論文投稿
論文を投稿する前後の話をすると、これはまぁなかなかしんどかったです。まず某P誌に投稿したのですが、投稿から2ヶ月経過しても返事が来ませんでした。痺れを切らせてHandling Editorに連絡したのですが、その直後にEditorが蒸発したという連絡がEditorial Officeから届きました。しかも、Editorial Office側は「あと2週間待って!今から査読者を用意して対応するから!」って話し始め、私も最初はそれに乗っかってしまったのですが、そこで2週間待った挙句にメールを出すと、往復のたびにEditorial Office側から「今やってるところです」→「あっ今査読者から『あと1週間待ってくれ』ってメッセージが来てます」→「あったった今コメントがHandling Editorから届きました」という小出しな引き止め行為をしはじめたので、さすがに呆れて「原稿をwithdrawさせんかい!!!!!」というメールを出そうとしたらChatGPTによって『その表現はちくちく言葉ですので丁寧にしてあげますね』と添削されたメールを代わりにお出ししたのですが、全然応じてくれず、100日経過してからようやく新Editorから「今査読終わったけど、本当にWithdrawする??今ならダッシュボードからwithdrawボタンを押せるやで」というメールが届きました。あまりに嫌気がさしたのでwithdrawを宣言し、他ジャーナルにようやく投稿することができました。なお、投稿してから知ったのですが、この少し前にP誌に投稿していた別の予知生合成科学の研究者もいらっしゃったのですが、その方も結局1回目の返事に3ヶ月、2回目にも3ヶ月かかったということを伺いました。もうこのジャーナルには投稿しまいと思いました。
しかし、そうこうしている間(2026年1〜3月)には関心を引くモデル生物種で似たような網羅的な解析を行った論文が先にNatureやNat. Commun.などから出てしまっていました。面白かったのは、2026年1月20日に出たMolecular Systems Biologyから出た論文で、これは113,050件のタンパク質ペア予測を行っているのですが、どうもこれはAlphaFold3公式のWebサーバーでずっと予測を回していたのではないかと思われます(!)。AlphaFold3は1つのGoogleアカウントで1日30件しか予測できないはずですが、いったいどうやって……??
それはさておき、とりあえずものは試しということでこれらのジャーナルに投稿してみたのですが、結局Nature系の姉妹誌には(投稿しなかったSci. Rep.を除いて)全部Editor rejectで落とされました。その中の1つのコメントは「その原稿は多くの読者に読んでもらえるような内容ではないから」という評価でした。やっぱりヒトや酵母みたいなメジャーな生物でやらないと載らないんですかね……と思ってしまいました(出るのが早かったのですが)。なお、それらの類似論文はAlphaFold3ではなくColabFoldを用いて大規模な予測を行っていました。ColabFoldの計算速度はAlphaFold2よりは10〜30倍ほど速いですが、AlphaFold3を用いた場合よりも5〜10倍以上は遅いです。それにも関わらず、今回の私の計算数(48万前後)よりも多い100万件以上を予測していたということから、おそらくColabFoldが実用的になった2022年くらいから2年以上くらいかけて、その間ColabFoldを我々の用いたGPU数よりもずっと多い数で回していたのかなと推察しています。また2026年3月にはNVIDIA社とColabFoldの開発者たちがColabFoldでUniProtに登録されている2億のタンパク質すべてについてホモダイマー予測を行ったという報告を行っています。NVIDIA社はGPUを事実上無限に使える環境を持っているので、我々のような大学の研究室では到底及ばない計算量で予測を行うことができるのだろうと思います。GPUパワーにはまいったな!!
結局、総合誌系は諦めて再度計算生物学の専門誌を検討した結果、Computational and Structural Biotechnology Journalに投稿することに決めました。こちらは結局Impact FactorはP誌とそんなに変わらないので、結果的にはP誌でそのまま粘ってたほうが良かったかもしれませんが、こちらは非常にEditorもReviewerも対応が早くて助かりました。CSBJ誌は2025年までElsevier管轄でしたが、今はAAASによって管理されているようです。私のようなAlphaFoldを用いた解析を行う研究者としてはありがたいのですが、なぜかReviewerが前回も今回も4人付いてくるのが大変です。
多くの生命科学系研究者の方にはなかなか通じないところかと思うのですが、今回のような内容のAlphaFoldを用いた計算的な構造生物学を査読してもらえるような論文誌というのは実は非常に限られておりまして、大型の総合誌(Nat. Commun.とか)でなければごく限られた計算系生物専門誌しか投稿するところがありません。加えて、最近ではAlphaFoldがあまりにも簡単にデータを出してくれるためか、bioRxivをはじめとしたプレプリントサーバー、もっと言えばあまり有名ではない計算系ジャーナルでさえ、毎日「AlphaFoldを使ってみた」系のプレプリントが投稿されるようになっているらしく、そうしたジャーナルの査読機構が機能不全に陥っている話を耳にするようになりました。いわゆる、機械学習・数学系プレプリントのarXivがまさに陥っている状況とほぼ同じです。先述のP誌のEditorial Officeもそうなっているのかもしれません。反対にNature Publishing GroupやElsevierなどの商業的、またはAAASのような非営利ながらハイブランドな大手出版社であればEditorが何人もついているでしょうから、今後はそういう大手ジャーナルだけが生き残り、学会と密接な関係にある古くからの学術専門誌は縮小していってしまうのではないかと危惧しています。
興味深いと思った点
共著者の尾瀬先生(北海道大学)・南先生(東京科学大学)・及川先生(北海道大学)たちのグループから2026年4月に公開されたMonBI-MonBIIキメラタンパク質構造の論文によって、MonBIは単体では酵素活性を持たないが(触媒活性を持つアミノ酸が消失している)、MonBIIを水溶液中で正しくフォールディングさせる働きがあることが示されています。面白いことに、MonBIは活性がないにも関わらずそれ自体でホモ二量体を形成し、結晶構造(PDB ID: 3WMD)も解かれています。つまり、MonBIは単体ではホモ二量体を形成しますが、MonBIIが近くにいるとヘテロ二量体を優先的に形成するようです。しかしながら、そもそもなぜそんなことをわざわざする必要があるのか?という酵素側の気持ちはまだまったくもって分かっていません。ただ言えるのは、AlphaFold3を用いることでそのような性質をもつタンパク質を検出することは(BGCの系以外でも)可能になりました。もしかすると他の生物種でもそうしたものが眠っているのかもしれません。2026年でもタンパク質のフォールディング機構や、常温でのタンパク質のダイナミクスを正確に予想することはとても困難です。今後はそうした協働するタンパク質のフォールディング機構の予測ができるようになると面白いかもしれないですね。
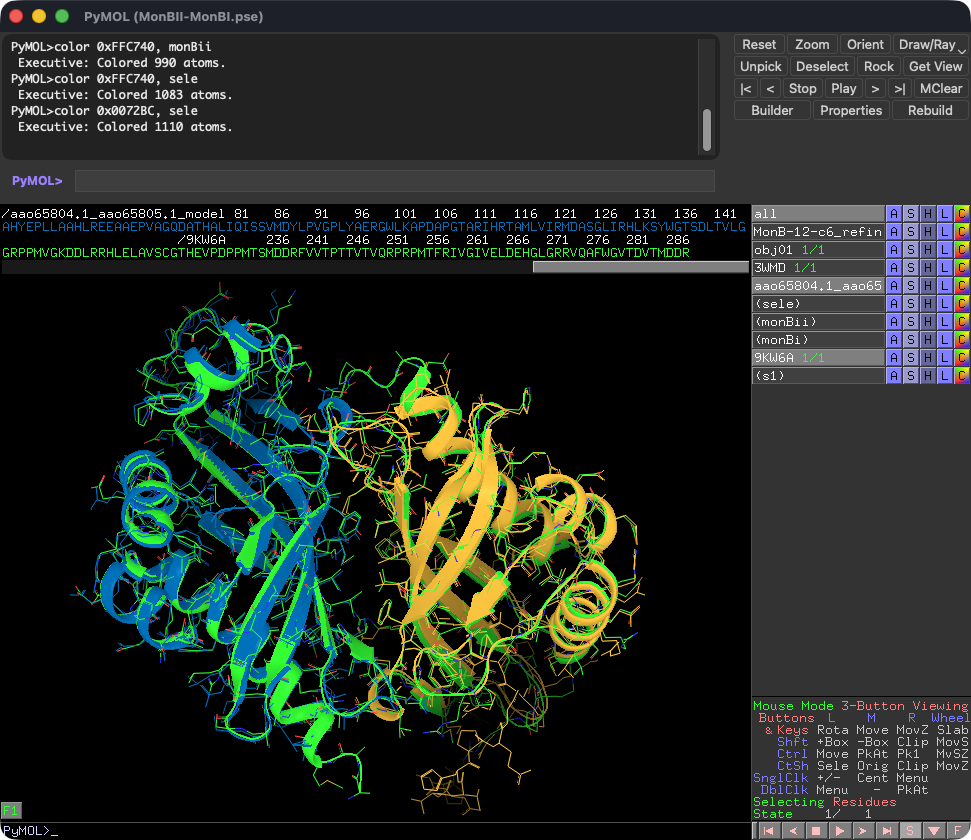
Figure. MonBI-MonBIIのAlphaFold3予測構造(MonBI:青, MonBII:黄)とキメラ体の結晶構造(PDB ID: 9KW6, 緑)の重ね合わせ
宣伝
計算創薬科学研究室では、本研究で紹介したような深層学習構造予測と分子シミュレーションの融合、あるいは創薬応用に興味のある大学院生を募集しています。ぜひ研究室のWebサイトをご覧いただくか、お問い合わせください。
本記事は2026年6月時点の研究成果に基づいています。
参考文献
[1] Zhang, S., Mukherji, R., Chowdhury, S., et al. Lipopeptide-mediated bacterial interaction enables cooperative predator defense, Proc. Natl. Acad. Sci. USA 118, e2013759118 (2021), DOI: 10.1073/pnas.2013759118.
[2] Chevrette, M., Thomas, C., Hurley, A., et al. Microbiome composition modulates secondary metabolism in a multispecies bacterial community, Proc. Natl. Acad. Sci. USA 119, e2212930119 (2022), DOI: 10.1073/pnas.2212930119.
[3] Modolon, F., Barno, A., Villela, H., et al. Ecological and biotechnological importance of secondary metabolites produced by coral-associated bacteria, J. Appl. Microbiol. 129, 1441–1457 (2020), DOI: 10.1111/jam.14766.
[4] Malpartida, F., and Hopwood, D. A. Physical and genetic characterisation of the gene cluster for the antibiotic actinorhodin in Streptomyces coelicolor A3(2), Mol. Genet. Genomics 205, 66–73 (1986),
[5] Stanzak, R., Matsushima, P., Baltz, R., et al. Cloning and Expression in Streptomyces lividans of Clustered Erythromycin Biosynthesis Genes from Streptomyces erythreus, Bio/Technology 4, 229–232 (1986), DOI: 10.1038/nbt0386-229.
[6] Medema, M., Blin, K., Cimermancic, P., et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences, Nucleic Acids Res. 39, W339–W346 (2011), DOI: 10.1093/nar/gkr466.
[7] Blin, K., Medema, M., Kazempour, D., et al. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers, Nucleic Acids Res. 41, W204–W212 (2013), DOI: 10.1093/nar/gkt449.
[8] Weber, T., Blin, K., Duddela, S., et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters, Nucleic Acids Res. 43, W237–W243 (2015), DOI: 10.1093/nar/gkv437.
[9] Blin, K., Wolf, T., Chevrette, M., et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification, Nucleic Acids Res. 45, W36–W41 (2017), DOI: 10.1093/nar/gkx319.
[10] Blin, K., Shaw, S., Steinke, K., et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline, Nucleic Acids Res. 47, W81–W87 (2019), DOI: 10.1093/nar/gkz310.
[11] Blin, K., Shaw, S., Kloosterman, A., et al. antiSMASH 6.0: improving cluster detection and comparison capabilities, Nucleic Acids Res. 49, W29–W35 (2021), DOI: 10.1093/nar/gkab335.
[12] Blin, K., Shaw, S., Augustijn, H., et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation, Nucleic Acids Res. 51, W46–W50 (2023), DOI: 10.1093/nar/gkad344.
[13] Blin, K., Shaw, S., Vader, L., et al. antiSMASH 8.0: extended gene cluster detection capabilities and analyses of chemistry, enzymology, and regulation, Nucleic Acids Res. 53, W32–W38 (2025), DOI: 10.1093/nar/gkaf334.
[14] Zdouc, M., and Blin, K., and Louwen, N., et al. MIBiG 4.0: advancing biosynthetic gene cluster curation through global collaboration, Nucleic Acids Res. 53, D678–D690 (2025), DOI: 10.1093/nar/gkae1115.
[15] Hannigan, G., Prihoda, D., Palicka, A., et al. A deep learning genome-mining strategy for biosynthetic gene cluster prediction, Nucleic Acids Res. 47 (2019), DOI: 10.1093/nar/gkz654.
[16] Cimermancic, P., Medema, M., Claesen, J., et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters, Cell 158, 412–421 (2014), DOI: 10.1016/j.cell.2014.06.034.
[17] Gulick, A., and Aldrich, C. Trapping interactions between catalytic domains and carrier proteins of modular biosynthetic enzymes with chemical probes, Nat. Prod. Rep. 35, 1156–1184 (2018), DOI: 10.1039/c8np00044a.
[18] Kosol, S., Jenner, M., Lewandowski, J., et al. Protein–protein interactions in trans-AT polyketide synthases, Nat. Prod. Rep. 35, 1097–1109 (2018), DOI: 10.1039/c8np00066b.
[19] Dodge, G., Maloney, F., and Smith, J. Protein–protein interactions in “cis-AT” polyketide synthases, Nat. Prod. Rep. 35, 1082–1096 (2018), DOI: 10.1039/c8np00058a.
[20] Herbst, D., Townsend, C., and Maier, T. The architectures of iterative type I PKS and FAS, Nat. Prod. Rep. 35, 1046–1069 (2018), DOI: 10.1039/c8np00039e.
[21] Chen, A., Re, R., and Burkart, M. Type II fatty acid and polyketide synthases: deciphering protein–protein and protein–substrate interactions, Nat. Prod. Rep. 35, 1029–1045 (2018), DOI: 10.1039/c8np00040a.
[22] Mori, T., Moriwaki, Y., Sakurada, K., et al. Molecular basis for the diversification of lincosamide biosynthesis by pyridoxal phosphate-dependent enzymes, Nat. Chem. 17, 256–264 (2025), DOI: 10.1038/s41557-024-01687-7.
[23] Sikandar, A., and Koehnke, J. The role of protein-protein interactions in the biosynthesis of ribosomally synthesized and post-translationally modified peptides, Nat. Prod. Rep. 36, 1576–1588 (2019), DOI: 10.1039/c8np00064f.
[24] Rattray, D., and Foster, L. Dynamics of protein complex components, Curr. Opin. Chem. Biol. 48, 81–85 (2019), DOI: 10.1016/j.cbpa.2018.11.003.
[25] Jumper, J., Evans, R., Pritzel, A., et al. Highly accurate protein structure prediction with AlphaFold, Nature 596, 583–589 (2021), DOI: 10.1038/s41586-021-03819-2.
[26] McCoy, A., Sammito, M., and Read, R. Implications of AlphaFold2 for crystallographic phasing by molecular replacement, Acta Crystallographica Section D: Structural Biology 78, 1–13 (2022), DOI: 10.1107/S2059798321012122.
[27] Akdel, M., Pires, D., Pardo, E., et al. A structural biology community assessment of AlphaFold2 applications, Nat. Struct. Mol. Biol. 29, 1056–1067 (2022), DOI: 10.1038/s41594-022-00849-w.
[28] Homma, M., Wakabayashi, T., Moriwaki, Y., et al. Insights into stereoselective ring formation in canonical strigolactone: Discovery of a dirigent domain-containing enzyme catalyzing orobanchol synthesis, Proc. Natl. Acad. Sci. USA 121, e2313683121 (2024), DOI: 10.1073/pnas.2313683121.
[29] Evans, R., O’Neill, M., Pritzel, A., et al. Protein complex prediction with AlphaFold-Multimer, bioRxiv, 2021.2010.2004.463034 (2022), DOI: 10.1101/2021.10.04.463034.
[30] Deneke, V., Blaha, A., Lu, Y., et al. A conserved fertilization complex bridges sperm and egg in vertebrates, Cell 187, 7066–7078.e7022 (2024), DOI: 10.1016/j.cell.2024.09.035.
[31] Abramson, J., Adler, J., Dunger, J., et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature 630, 493–500 (2024), DOI: 10.1038/s41586-024-07487-w.
[32] Eddy, S. R. Accelerated Profile HMM Searches, PLoS Comp. Biol. 7, e1002195 (2011), DOI: 10.1371/journal.pcbi.1002195.
[33] Mirdita, M., Schütze, K., Moriwaki, Y., et al. ColabFold: making protein folding accessible to all, Nat. Methods 19, 679–682 (2022), DOI: 10.1038/s41592-022-01488-1.
[34] Steinegger, M., and Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets, Nat. Biotechnol. 35, 1026–1028 (2017), DOI: 10.1038/nbt.3988.
[35] Dunbrack, R. L. Rēs ipSAE loquunt: What’s wrong with AlphaFold’s ipTM score and how to fix it, bioRxiv, 2025.2002.2010.637595 (2025), DOI: 10.1101/2025.02.10.637595.
[36] Overath, M. D., Rygaard, A. S. H., Jacobsen, C. P., et al. Predicting Experimental Success in De Novo Binder Design: A Meta-Analysis of 3,766 Experimentally Characterised Binders, bioRxiv, 2025.2008.2014.670059 (2025), DOI: 10.1101/2025.08.14.670059.
[37] Bryant, P., Pozzati, G., and Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2, Nat. Commun. 13, 1265 (2022), DOI: 10.1038/s41467-022-28865-w.
[38] Zhu, W., Shenoy, A., Kundrotas, P., et al. Evaluation of AlphaFold-Multimer prediction on multi-chain protein complexes, Bioinformatics 39, btad424 (2023), DOI: 10.1093/bioinformatics/btad424.
[39] Kim, A.-R., Hu, Y., Comjean, A., et al. Enhanced Protein-Protein Interaction Discovery via AlphaFold-Multimer, bioRxiv, 2024.2002.2019.580970 (2024), DOI: 10.1101/2024.02.19.580970.
[40] Wojdyr, M. GEMMI: A library for structural biology, J. Open Source Softw. 7, 4200 (2022), DOI: 10.21105/joss.04200.
[41] Zhang, J., Yuzawa, S., Thong, W., et al. Reconstitution of a Highly Reducing Type II PKS System Reveals 6π-Electrocyclization Is Required for o-Dialkylbenzene Biosynthesis, J. Am. Chem. Soc. 143, 2962–2969 (2021), DOI: 10.1021/jacs.0c13378.
[42] Krissinel, E., and Henrick, K. Inference of macromolecular assemblies from crystalline state, J. Mol. Biol. 372, 774–797 (2007), DOI: 10.1016/j.jmb.2007.05.022.
[43] Barajas, J., Phelan, R., Schaub, A., et al. Comprehensive Structural and Biochemical Analysis of the Terminal Myxalamid Reductase Domain for the Engineered Production of Primary Alcohols, Chem. Biol. 22, 1018–1029 (2015), DOI: 10.1016/j.chembiol.2015.06.022.
[44] Wang, L., Parnell, A., Williams, C., et al. A Rieske oxygenase/epoxide hydrolase-catalysed reaction cascade creates oxygen heterocycles in mupirocin biosynthesis, Nat. Catal. 1, 968–976 (2018), DOI: 10.1038/s41929-018-0183-5.
[45] Miyanaga, A., Kurihara, S., Chisuga, T., et al. Structural Characterization of Complex of Adenylation Domain and Carrier Protein by Using Pantetheine Cross-Linking Probe, ACS Chem. Biol. 15, 1808–1812 (2020), DOI: 10.1021/acschembio.0c00403.
[46] Mori, T., Kadlcik, S., Lyu, S., et al. Molecular basis for carrier protein-dependent amide bond formation in the biosynthesis of lincosamide antibiotics, Nat. Catal. 6, 531–542 (2023), DOI: 10.1038/s41929-023-00971-y.
[47] Miyanaga, A., Nagata, K., Nakajima, J., et al. Structural Basis of Amide-Forming Adenylation Enzyme VinM in Vicenistatin Biosynthesis, ACS Chem. Biol. 18, 2343–2348 (2023), DOI: 10.1021/acschembio.3c00517.
[48] Haslinger, K., Peschke, M., Brieke, C., et al. X-domain of peptide synthetases recruits oxygenases crucial for glycopeptide biosynthesis, Nature 521, 105–109 (2015), DOI: 10.1038/nature14141.
[49] Schramma, K. R., Bushin, L. B., and Seyedsayamdost, M. R. Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink, Nat. Chem. 7, 431–437 (2015), DOI: 10.1038/nchem.2237.
[50] Ortega, M. A., Hao, Y., Zhang, Q., et al. Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB, Nature 517, 509–512 (2015), DOI: 10.1038/nature13888.
[51] Bothwell, I. R., Cogan, D. P., Kim, T., et al. Characterization of glutamyl-tRNA-dependent dehydratases using nonreactive substrate mimics, Proc. Natl. Acad. Sci. USA 116, 17245–17250 (2019), DOI: 10.1073/pnas.1905240116.
[52] Davis, K. M., Schramma, K. R., Hansen, W. A., et al. Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition, Proc. Natl. Acad. Sci. USA 114, 10420–10425 (2017), DOI: 10.1073/pnas.1703663114.
[53] Olano, C., Wilkinson, B., Moss, S., et al. Evidence from engineered gene fusions for the repeated use of a module in a modular polyketide synthase, Chem. Commun., 2780–2782 (2003), DOI: 10.1039/b310648a.
[54] Olano, C., Moss, S., Braña, A., et al. Biosynthesis of the angiogenesis inhibitor borrelidin by Streptomyces parvulus Tü4055: insights into nitrile formation, Mol. Microbiol. 52, 1745–1756 (2004), DOI: 10.1111/j.1365-2958.2004.04090.x.
[55] Olano, C., Wilkinson, B., Sánchez, C., et al. Biosynthesis of the angiogenesis inhibitor borrelidin by Streptomyces parvulus Tü4055: Cluster analysis and assignment of functions, Chem. Biol. 11, 87–97 (2004), DOI: 10.1016/j.chembiol.2003.12.018.
[56] Camacho, C., Coulouris, G., Avagyan, V., et al. BLAST+ : architecture and applications, BMC Bioinformatics 10, 421 (2009), DOI: 10.1186/1471-2105-10-421.
[57] van Kempen, M., Kim, S. S., Tumescheit, C., et al. Fast and accurate protein structure search with Foldseek, Nat. Biotechnol. 42, 243–246 (2024), DOI: 10.1038/s41587-023-01773-0.
[58] Grunwald, S., Hopf, L., Bock-Bierbaum, T., et al. Divergent architecture of the heterotrimeric NatC complex explains N-terminal acetylation of cognate substrates, Nat. Commun. 11, 5506 (2020), DOI: 10.1038/s41467-020-19321-8.
[59] Hamed, A., Abdel-Razek, A., Frese, M., et al. N-Acetylborrelidin B: a new bioactive metabolite from Streptomyces mutabilis sp MII, Z. Naturforsch., C: Biosci. 73, 49–57 (2018), DOI: 10.1515/znc-2017-0140.
[60] Varadi, M., Anyango, S., Deshpande, M., et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res. 50, D439–D444 (2022), DOI: 10.1093/nar/gkab1061.
[61] Brock, N., Nikolay, A., and Dickschat, J. Biosynthesis of the antibiotic tropodithietic acid by the marine bacterium Phaeobacter inhibens, Chem. Commun. 50, 5487–5489 (2014), DOI: 10.1039/c4cc01924e.
[62] Keatinge-Clay, A., Maltby, D., Medzihradszky, K., et al. An antibiotic factory caught in action, Nat. Struct. Mol. Biol. 11, 888–893 (2004), DOI: 10.1038/nsmb808.
[63] Deng, Z., Liu, J., Li, T., et al. An Unusual Type II Polyketide Synthase System Involved in Cinnamoyl Lipid Biosynthesis, Angew. Chem. Int. Ed. 60, 153–158 (2021), DOI: 10.1002/anie.202007777.
[64] Minami, A., Ose, T., Sato, K., et al. Allosteric Regulation of Epoxide Opening Cascades by a Pair of Epoxide Hydrolases in Monensin Biosynthesis, ACS Chem. Biol. 9, 562–569 (2014), DOI: 10.1021/cb4006485.
[65] Yabuno, N., Minami, A., Ozaki, T., et al. A system of paired polyether epoxide hydrolases enables a mouldable enzyme for consecutive ring cyclization cascades, Nat. Chem. (2026), DOI: 10.1038/s41557-026-02122-9.
[66] Fu, C., Xie, F., Hoffmann, J., et al. Armeniaspirol Antibiotic Biosynthesis: Chlorination and Oxidative Dechlorination Steps Affording Spiro[4.4]non-8-ene, ChemBioChem 20, 764–769 (2019), DOI: 10.1002/cbic.201800791.
[67] Pang, A., Garneau-Tsodikova, S., and Tsodikov, O. Crystal structure of halogenase PltA from the pyoluteorin biosynthetic pathway, J. Struct. Chem. 192, 349–357 (2015), DOI: 10.1016/j.jsb.2015.09.013.
[68] Barker, R. D., Yu, Y., De Maria, L., et al. Mechanism of Action of Flavin-Dependent Halogenases, ACS Catal. 12, 15352–15360 (2022), DOI: 10.1021/acscatal.2c05231.
[69] Yeh, E., Blasiak, L. C., Koglin, A., et al. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases, Biochemistry 46, 1284–1292 (2007), DOI: 10.1021/bi0621213.
[70] Flecks, S., Patallo, E. P., Zhu, X., et al. New insights into the mechanism of enzymatic chlorination of tryptophan, Angew. Chem. Int. Ed. 47, 9533–9536 (2008), DOI: 10.1002/anie.200802466.
[71] Remmert, M., Biegert, A., Hauser, A., et al. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment, Nat. Methods 9, 173–175 (2012), DOI: 10.1038/nmeth.1818.
[72] Kallenborn, F., Chacon, A., Hundt, C., et al. GPU-accelerated homology search with MMseqs2, Nat. Methods 22, 2024–2027 (2025), DOI: 10.1038/s41592-025-02819-8.
[73] Perry, B. C., Kim, J., and Romero, P. A. AlphaFast: High-throughput AlphaFold 3 via GPU-accelerated MSA construction, bioRxiv, 2026.2002.2017.706409 (2026), DOI: 10.64898/2026.02.17.706409.
[74] Ames, B., Nguyen, C., Bruegger, J., et al. Crystal structure and biochemical studies of the trans-acting polyketide enoyl reductase LovC from lovastatin biosynthesis, Proc. Natl. Acad. Sci. USA 109, 11144–11149 (2012), DOI: 10.1073/pnas.1113029109.
[75] Matthews, M., Krest, C., Barr, E., et al. Substrate-Triggered Formation and Remarkable Stability of the C-H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase, SyrB2, Biochemistry 48, 4331–4343 (2009), DOI: 10.1021/bi900109z.
[76] Li, X., Awakawa, T., Mori, T., et al. Heterodimeric Non-heme Iron Enzymes in Fungal Meroterpenoid Biosynthesis, J. Am. Chem. Soc. 143, 21425–21432 (2021), DOI: 10.1021/jacs.1c11548.
[77] del Alamo, D., Sala, D., McHaourab, H. S., et al. Sampling alternative conformational states of transporters and receptors with AlphaFold2, eLife 11 (2022), DOI: 10.7554/eLife.75751.
[78] Giovannoni, S., Tripp, H., Givan, S., et al. Genome streamlining in a cosmopolitan oceanic bacterium, Science 309, 1242–1245 (2005), DOI: 10.1126/science.1114057.
[79] Giovannoni, S., Thrash, J., and Temperton, B. Implications of streamlining theory for microbial ecology, ISME J. 8, 1553–1565 (2014), DOI: 10.1038/ismej.2014.60.
[80] Kotaka, M., Ren, J., Lockyer, M., et al. Structures of R- and T-state Escherichia coli aspartokinase III: Mechanisms of the allosteric transition and inhibition by lysine, J. Biol. Chem. 281, 31544–31552 (2006), DOI: 10.1074/jbc.M605886200.
[81] Mattevi, A., Valentini, G., Rizzi, M., et al. Crystal structure of Escherichia coli pyruvate kinase type I: molecular basis of the allosteric transition, Structure 3, 729–741 (1995), DOI: 10.1016/S0969-2126(01)00207-6.
[82] Zhang, D., Zhang, F., and Liu, W. A KAS-III Heterodimer in Lipstatin Biosynthesis Nondecarboxylatively Condenses C8 and C14 Fatty Acyl-CoA Substrates by a Variable Mechanism during the Establishment of a C22 Aliphatic Skeleton, J. Am. Chem. Soc. 141, 3993–4001 (2019), DOI: 10.1021/jacs.8b12843.
[83] Randall, G., Grant-Mackie, E., Chunkath, S., et al. A Stable Dehydratase Complex Catalyzes the Formation of Dehydrated Amino Acids in a Class V Lanthipeptide, ACS Chem. Biol. 19, 2548–2556 (2024), DOI: 10.1021/acschembio.4c00637.
[84] Ren, M., Li, Z., Wang, Z., et al. Antiviral Chlorinated Drimane Meroterpenoids from the Fungus Talaromyces pinophilus LD-7 and Their Biosynthetic Pathway, J. Nat. Prod. 87, 2034–2044 (2024), DOI: 10.1021/acs.jnatprod.4c00539.
[85] Hotta, K., Chen, X., Paton, R., et al. Enzymatic catalysis of anti-Baldwin ring closure in polyether biosynthesis, Nature 483, 355–358 (2012), DOI: 10.1038/nature10865.
[86] Minami, A., Oguri, H., Watanabe, K., et al. Biosynthetic machinery of ionophore polyether lasalocid: enzymatic construction of polyether skeleton, Curr. Opin. Chem. Biol. 17, 555–561 (2013), DOI: 10.1016/j.cbpa.2013.06.004.
[87] Migita, A., Watanabe, M., Hirose, Y., et al. Identification of a Gene Cluster of Polyether Antibiotic Lasalocid from Streptomyces lasaliensis, Biosci. Biotechnol. Biochem. 73, 169–176 (2009), DOI: 10.1271/bbb.80631.
[88] Wong, F., Hotta, K., Chen, X., et al. Epoxide Hydrolase-Lasalocid A Structure Provides Mechanistic Insight into Polyether Natural Product Biosynthesis, J. Am. Chem. Soc. 137, 86–89 (2015), DOI: 10.1021/ja511374k.
[89] Shi, J., Shi, Y., Li, J., et al. In Vitro Reconstitution of Cinnamoyl Moiety Reveals Two Distinct Cyclases for Benzene Ring Formation, J. Am. Chem. Soc. 144, 7939–7948 (2022), DOI: 10.1021/jacs.2c02855.
[90] Shi, J., Liu, C., Zhang, B., et al. Genome mining and biosynthesis of kitacinnamycins as a STING activator, Chem. Sci. 10, 4839–4846 (2019), DOI: 10.1039/c9sc00815b.
[91] Dong, C., Flecks, S., Unversucht, S., et al. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination, Science 309, 2216–2219 (2005), DOI: 10.1126/science.1116510.
[92] Moynié, L., Serra, I., Scorciapino, M., et al. Preacinetobactin not acinetobactin is essential for iron uptake by the BauA transporter of the pathogen Acinetobacter baumannii, eLife 7, e42270 (2018), DOI: 10.7554/eLife.42270.
[93] Zhu, J., Lippa, G., Gulick, A., et al. Examining Reaction Specificity in PvcB, a Source of Diversity in Isonitrile-Containing Natural Products, Biochemistry 54, 2659–2669 (2015), DOI: 10.1021/acs.biochem.5b00255.
[94] Fenwick, M., Philmus, B., Begley, T., et al. Burkholderia glumae ToxA Is a Dual-Specificity Methyltransferase That Catalyzes the Last Two Steps of Toxoflavin Biosynthesis, Biochemistry 55, 2748–2759 (2016), DOI: 10.1021/acs.biochem.6b00167.
[95] Chi, C., Wang, Z., Liu, T., et al. Crystal Structures of Fsa2 and Phm7 Catalyzing [4+2] Cycloaddition Reactions with Reverse Stereoselectivities in Equisetin and Phomasetin Biosynthesis, ACS Omega 6, 12913–12922 (2021), DOI: 10.1021/acsomega.1c01593.
[96] Keating, T., Marshall, C., Walsh, C., et al. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains, Nat. Struct. Biol. 9, 522–526 (2002), DOI: 10.1038/nsb810.