Last year, we published a paper in ACS Omega [1] introducing a restraint-guided inference method that improves ligand stereochemistry on top of Boltz-1. More than half a year has passed since then, and the field has moved quickly: not only has Boltz-2 [2] arrived, but newer models such as Protenix-v1/v2 [3][4] and OpenFold3 [6] have been released in rapid succession. In this post, we re-run our benchmark against these latest models to see how much the ligand-stereochemistry issues we flagged previously have actually improved.
Benchmark conditions
We compared the following six conditions.
- AlphaFold3
- Boltz-2 (vanilla)
- Boltz-2 + conformer restraints (our method applied)
- Boltz-2 w/ potential (using Boltz-2’s built-in inference-time potentials [2])
- Protenix-v1
- Protenix-v2
The evaluation dataset is the same PLINDER-based chiral-compound set [5] as in our previous paper. A note on training cutoff dates: Protenix-v1/v2 use the same cutoff as AF3 (September 30, 2021), so a Before/After split is meaningful as a generalization test. Boltz-2, on the other hand, has a much later cutoff of June 1, 2023, which does not line up with our Before/After split. This means that even if Boltz-2 looks strong on the “After” portion, some of that apparent strength may simply come from structures present in its training data, and the comparison is not strictly apples-to-apples. Please keep this caveat in mind.
For Boltz-2 + conformer restraints, we newly implemented the restraint-guided inference method for Boltz-2 in a dedicated repository (https://github.com/cddlab/boltz_restr), and the benchmark numbers are based on this code. This implementation adds structural guidance via distance restraints, as well as ligand–protein (VdW) restraints.
We have not included OpenFold3 [6] in this round of benchmarking. With the March 2026 preview2 update the training data has been fully released, making it a highly interesting project from the perspectives of reproducibility and extensibility, but for various practical reasons we could not include it this time. We hope to add it in a future benchmark.
Success rate
We first checked whether structure prediction completed normally and whether the ligand geometry remained intact (no broken bonds, no topology changes, etc.). The success rate (%) is the fraction of successful entries over the total.
| Model | Success rate (%) |
|---|---|
| AlphaFold3 | 99.86 |
| Boltz-2 w/ potential | 99.67 |
| Boltz-2 | 99.50 |
| Boltz-2 + conf restr | 99.62 |
| Protenix-v1 | 99.12 |
| Protenix-v2 | 94.73 |
Most models achieved over 99%, but Protenix-v2 produced broken topologies (ligand bonds stretching too far, etc.) in roughly 5% of cases, which is a bit concerning.
Chirality recovery
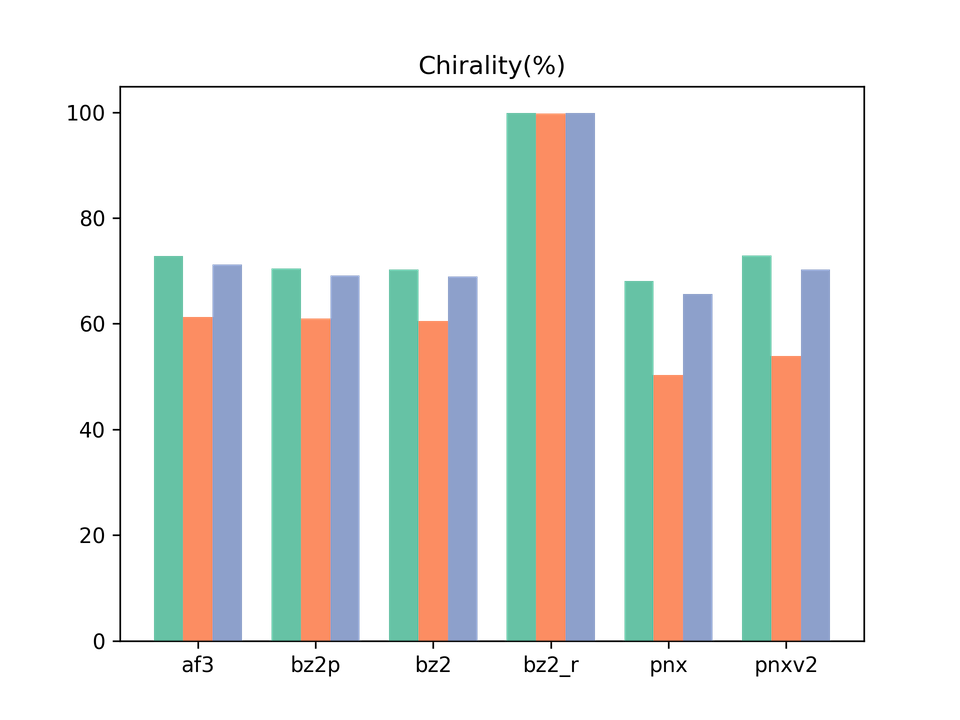
Legend: af3 = AlphaFold3, bz2p = Boltz-2 w/ potential, bz2 = Boltz-2 (vanilla), bz2_r = Boltz-2 + conformer restraints, pnx = Protenix-v1, pnxv2 = Protenix-v2. The three bars in each group correspond to Before / After / All.
This is one of the key metrics highlighted in our previous paper. The result: only Boltz-2 + conformer restraints achieved 100% chirality recovery. This is consistent with what we observed previously on Boltz-1, confirming that our method remains effective on newer backbones.
In contrast, Boltz-2’s inference-time potentials did not meaningfully improve chirality. Whether the potentials are actually working as intended, or whether additional settings are needed, remains to be verified.
A somewhat surprising result is that Protenix-v2 is slightly worse than v1 on chirality recovery. Despite substantial overall improvements to the model, this particular aspect does not seem to have progressed.
Protein structure accuracy (Protein RMSD)
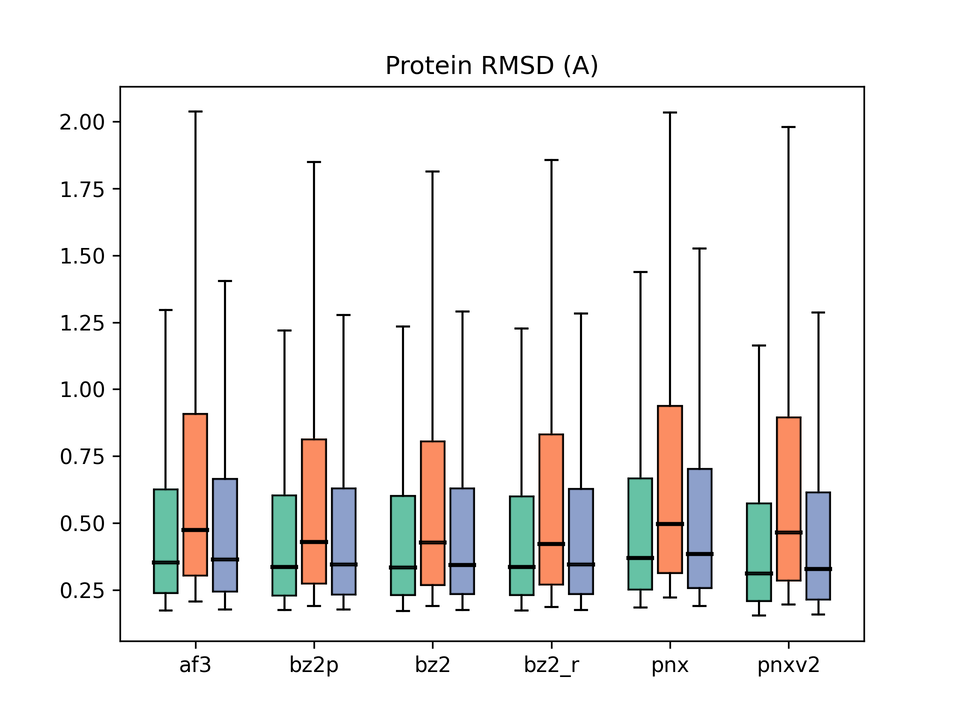
Legend: af3 = AlphaFold3, bz2p = Boltz-2 w/ potential, bz2 = Boltz-2 (vanilla), bz2_r = Boltz-2 + conformer restraints, pnx = Protenix-v1, pnxv2 = Protenix-v2. The three box plots in each group correspond to Before / After / All.
While not the primary focus of this benchmark, we also evaluated protein structure accuracy for reference. Boltz-2 showed the lowest median RMSD, with a small Before/After gap. Protenix-v2 improved over v1, but with a somewhat larger Before/After gap, suggesting a mild tendency toward overfitting. That said, all models produced acceptably accurate structures overall, and although there are visible differences, they are small and it is unclear whether any are statistically significant.
Ligand binding pose accuracy (Ligand RMSD)
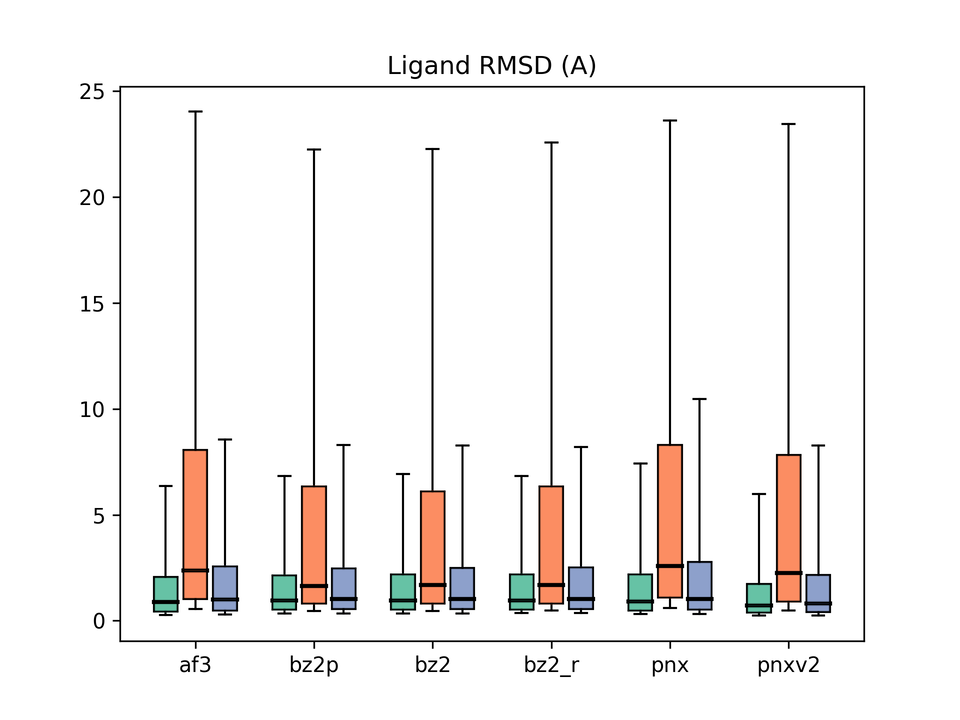
Legend: af3 = AlphaFold3, bz2p = Boltz-2 w/ potential, bz2 = Boltz-2 (vanilla), bz2_r = Boltz-2 + conformer restraints, pnx = Protenix-v1, pnxv2 = Protenix-v2. The three box plots in each group correspond to Before / After / All.
Next, ligand binding-pose prediction accuracy. On the All and Before (pre-cutoff) subsets, every model performs reasonably well, but differences emerge on the After (post-cutoff) subset.
Boltz-2 shows the best median Ligand RMSD. As noted above, however, this may in part reflect its later training cutoff. AF3 is, somewhat unexpectedly, not particularly strong here. Protenix-v1 scores the worst, but v2 shows clear improvement. On the other hand, the After/Before gap widens for v2, again hinting at overfitting.
Ligand geometry accuracy (Bond RMSD, Angle RMSD)
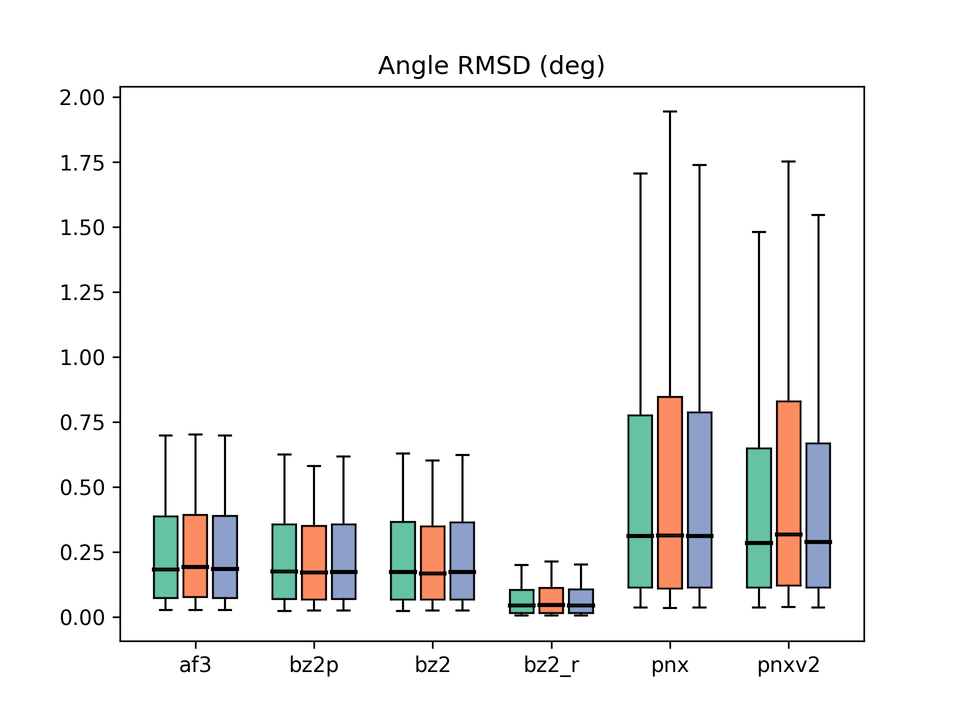
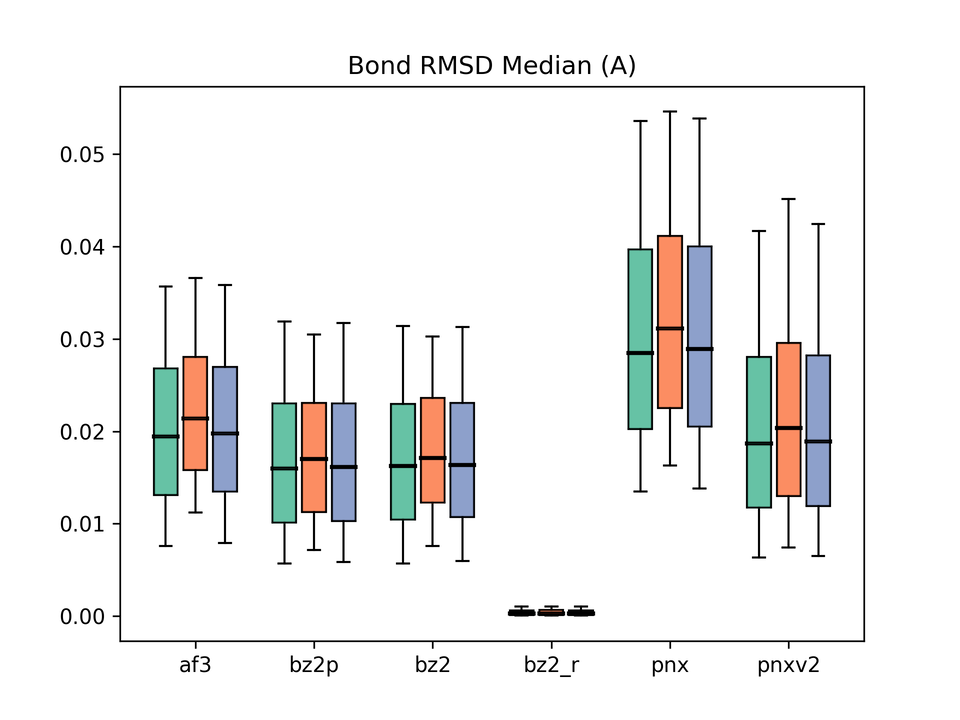
Legend: af3 = AlphaFold3, bz2p = Boltz-2 w/ potential, bz2 = Boltz-2 (vanilla), bz2_r = Boltz-2 + conformer restraints, pnx = Protenix-v1, pnxv2 = Protenix-v2. The three box plots in each group correspond to Before / After / All.
Another metric we emphasized in the previous paper. On bond-length and bond-angle accuracy, Boltz-2 again produced the best results. Enabling Boltz-2’s potentials barely moved these metrics, again raising questions about whether the potentials are exerting the intended effect.
In contrast, applying conformer restraints yields dramatic improvements, mirroring what we observed with Boltz-1. The effectiveness of restraint-guided inference is reconfirmed.
The Protenix family appears to have systematic issues with ligand geometry. Interestingly, v2 improves Bond RMSD but Angle RMSD is largely unchanged from v1. Why this asymmetry arises is an interesting question in its own right.
Minimum protein–ligand atom distance
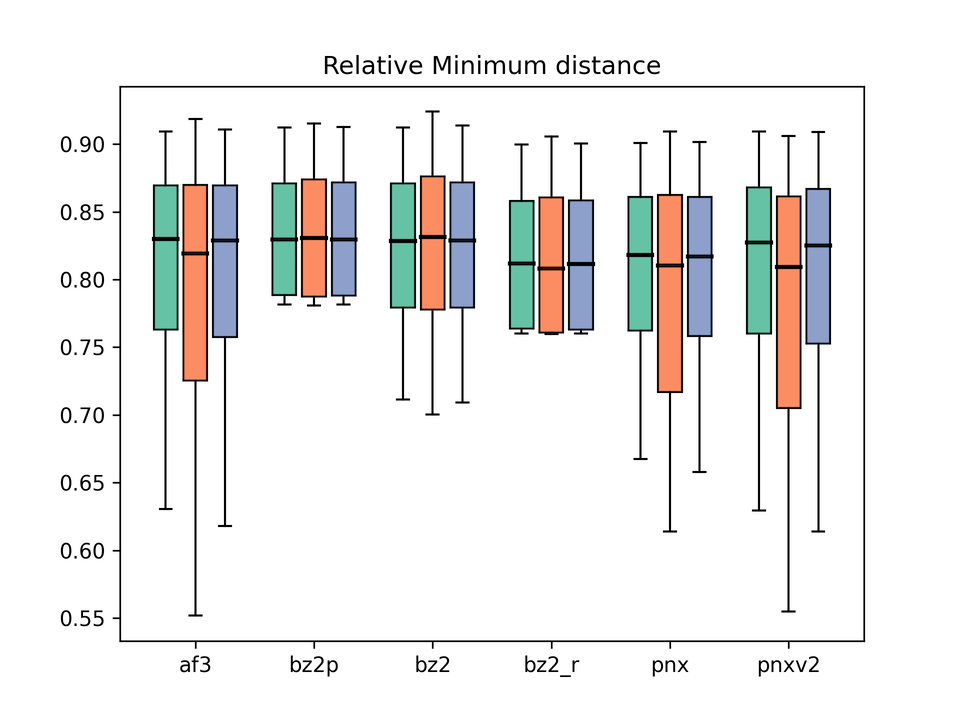
Legend: af3 = AlphaFold3, bz2p = Boltz-2 w/ potential, bz2 = Boltz-2 (vanilla), bz2_r = Boltz-2 + conformer restraints, pnx = Protenix-v1, pnxv2 = Protenix-v2. The three box plots in each group correspond to Before / After / All.
This metric assesses whether protein and ligand physically overlap (i.e., steric clashes). We use the PoseBusters-style relative inter-atomic distance — the distance normalized by the sum of VdW radii — and plot its minimum over the predicted structure. A value of 1 corresponds to roughly the VdW contact distance; values below 1 are possible when, for example, hydrogen bonds are being formed. As a rough rule of thumb, values below around 0.7 indicate atoms that are too close (clashing).
Looking at the results, Boltz-2 is already quite good in its vanilla form (bz2), and enabling the potentials (bz2p) essentially eliminates clashes. On this axis the potentials clearly have the intended effect. Boltz-2 with conformer restraints (bz2_r) also shows near-zero clash, confirming that the VdW restraints are working as designed.
By contrast, AF3 produces a fair number of steric clashes, and Protenix-v1/v2 similarly suffer from many overlaps, with no visible improvement in v2.
Summary and outlook
Key takeaways from this benchmark:
Overall, Boltz-2 produced the strongest results. Taken at face value, for applications such as cofolding, Boltz-2 (+ inference-time potentials / conformer restraints) appears to be the current best choice. As noted, however, the training-cutoff difference means this is not a purely performance-only comparison: some of Boltz-2’s apparent edge likely comes from having seen newer structures during training.
For chirality recovery, applying conformer restraints remains the most effective approach.
Boltz-2’s inference-time potentials (--use_potentials option) do help resolve steric clashes,
but contribute little to chirality or ligand-geometry improvements.
This deserves further investigation.
Protenix-v2, meanwhile, shows steady improvements. As noted in the authors’ technical report [4], it is clearly better than v1, though how it compares to AF3 and Boltz-2 in absolute terms will require more careful study.
One thing we did not explore this round: Protenix-v2 ships with a Training-Free Guidance (TFG) module [4] that allows geometric and physical constraints to be applied at inference time. We couldn’t get a clear enough handle on its usage to include it here, but it sounds conceptually similar to our conformer restraints, and we would like to try it out in a future post.
This post reflects the state of the field as of April 2026.
References
[1] Ishitani, R.; Moriwaki, Y. Improving Stereochemical Limitations in Protein–Ligand Complex Structure Prediction. ACS Omega 2025, 10, 45, 43857–43869.
[2] Passaro, S.; et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv 2025, 2025.06.14.659707.
[3] ByteDance AML AI4Science Team; et al. Protenix — Advancing Structure Prediction Through a Comprehensive AlphaFold3 Reproduction. bioRxiv 2025, 2025.01.08.631967.
[4] ByteDance AML AI4Science Team; et al. Protenix-v2: Broadening the Reach of Structure Prediction and Biomolecular Design. bioRxiv 2026, 2026.04.10.717613.
[5] Durairaj, J.; et al. PLINDER: The Protein–Ligand Interactions Dataset and Evaluation Resource. bioRxiv 2024, 2024.07.17.603955.
[6] The OpenFold3 Team. OpenFold3-preview. GitHub, 2025. https://github.com/aqlaboratory/openfold-3